Forward Documentation¶
Forward is a bioinformatics utility to facilitate phenomic studies using genetic cohorts (i.e it was not designed for pheWAS studies based on electronic medical records). It was built with a strong emphasis on flexibility, performance and reproducibility. The documented interfaces make it easy for bioinformaticians to extend Forward‘s capabilities by writing their own implementations (e.g. to add support for a new file format or to optimize computation for their dataset) and the automatic reporting and archiving functionality greatly facilitate the dissemination and reproduction of results.
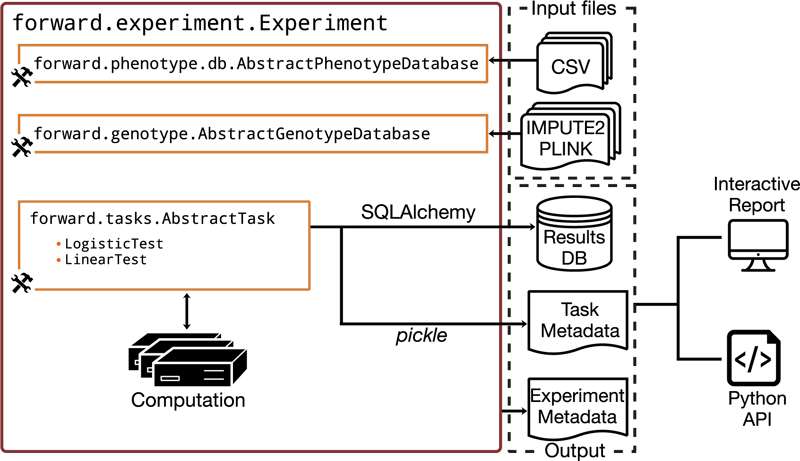
The different components of Forward. The customizable components have the tool icon.
Contents¶
Installation¶
To install Forward, simply run:
pip install forward
You can then run the tests using:
python -c 'import forward; forward.test()'
# Or if you want more verbosity:
python -c 'import forward; forward.test(2)'
Quick start¶
Configuration files¶
The easiest way to run a Forward experiment is to use the support for YAML configuration files. These files contain all the necessary information to define all the aspects of the experiment and serve as an archive to define what analysis was executed. They are automatically added to the interactive report.
An example can be found here:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 | Database:
pyclass: ExcelPhenotypeDatabase
missing_values: ["-9", "-99", "-88", "-77"]
sample_column: "Sample"
filename: /path/to/cohort_phenotypes.xlsx
exclude_correlated: 0.8
Variables:
- name: MyocInfarction
type: discrete
- name: BMI
type: continuous
transformation: log
- name: BPSystolic
type: continuous
- name: PC1
type: continuous
covariate: Yes
- name: PC2
type: continuous
covariate: Yes
- name: GenderFemale
type: discrete
covariate: Yes
Genotypes:
pyclass: MemoryImpute2Geno
filename: /path/to/data/impute2_extractor.impute2
samples: /path/to/data/forward_samples.txt
filter_probability: 0.90
filter_completion: 0.95
filter_maf: 0.01
filter_name: /path/to/data/variants.good_sites
exclude_samples: ["9210", ]
Experiment:
name: "ADCY9_forward"
cpu: 4
build: "GRCh37"
tasks:
- pyclass: LogisticTest
outcomes: all
covariates: all
- pyclass: LinearTest
outcomes: all
covariates: all
|
The Database block (lines 1-6) represents the phenotype database that is used
in the experiment. Multiple different pyclasses are availble to handle flat
files or Excel files, but implementations of the phenotype database interface
make it easy to extend this to other formats or databases. Some other options
are also included in this example, such as the missing_values command that
is passed to the underlying Python object to make sure that exclusions are
properly represented. The sample_column directive which column contains
sample IDs, filename is the path to the file containing phenotypes and
exclude_correlated is used to mark the correlation threshold to exclude
affected samples from the control groups of correlated outcomes. As an example
of this last command, if angina and myocardial infarction are correlated at
0.8, individuals with angina will be excluded from the control group for
myocardial infarction and the other way around.
The Variables block (lines 8-29) defines all the variables under study,
including covariates. The name command should correspond to phenotype IDs
from the Database section, the type command is used to identify continuous
and discrete variables and the transformation command can be used to
transform continuous traits (e.g. to achieve normality if the chosen
statistical tests requires it).
The Genotypes block (lines 31-40) is used to represent genotypic data. For
now, the included implementations of the genotype database interface are for
micrarray data in plink binary format or for imputed IMPUTE2 files. Basic
filtering of variants by imputation probability (filter_probability), by
completion rate (percentage of non-missing genotypes for a given marker,
filter_completion), by minor allele frequency (filter_maf) and by
variant name (using a file with a single columns, filter_name) is also
built-in for the MemoryImpute2Geno class. Note that the Memory in the
class name is because everything will be loaded in memory, which means that it
will be fast for small genetic datasets, but it won’t be suitable for larger
datasets. Users are encouraged to either extract their region of interest or to
implement a version that does supports indexing on the hard disk.
The Experiment block (lines 42-54) defines all the analyses that will be
executed by Forward. The name is used as an identifier and the corresponding
folder will be automatically created. If it already exists, Forward will
refuse to run (because we don’t want to overwrite your data). The cpu
instruction will be passed to the tasks and will determine how many parallel
processes will be ran (if supported for the chosen tasks). The build
(e.g. GRCh37) will be archived with other meta information to ensure
reproductibility and could eventually be used in the interactive report.
The tasks list is to tell Forward what statistical analyses are to be
executed as part of this experiment. For now, only methods for common variant
association testing are available (linear and logistic regression), but we
are actively working on other statistical tests.
Running an experiment¶
To run the newly created configuration file, you can use the command line
interface script (forward/scripts/forward-cli.py). Eventually, this will be
automatically installed. The usage is simple, just pass the path to the
configuration file.
forward-cli run my_configuration.yaml
A sample outpout will then look like:
INFO:forward.genotype:Loading samples from data/impute2/forward_samples.txt
INFO:forward.genotype:Setting the MAF threshold to 0.01
INFO:forward.genotype:Setting the completion threshold to 0.95
INFO:forward.genotype:Keeping only variants with IDs in file: 'data/impute2/chr16.imputed.good_sites'
INFO:forward.experiment:The build set for this experiment is GRCh37.
WARNING:forward.phenotype.db:Some samples were discarded when reordering phenotype information (1343 samples discarded). This could be because no genotype information is available for these samples.
INFO:forward.genotype:Built the variant database (17 entries).
INFO:root:Running a logistic regression analysis.
INFO:root:Running a linear regression analysis.
INFO:forward.experiment:Completed all tasks in 00:00:51.
To view the interactive report, use the forward-cli script:
forward-cli report my_experiment
To view the generated interactive report, you can then follow the on-screen instructions:
forward-cli report my_experiment
and go to http://127.0.0.1:5000/forward with your favorite browser.
A sample report is available on StatGen’s website.
Also note that the interactive report is strictly optional and you can still browse the analyses results manually. See the next section for details.
Browsing results¶
After executing an experiment, the following directory structure will have been created:
└── My_Experiment
├── configuration.yaml
├── experiment_info.pkl
├── forward_database.db
├── phen_correlation_matrix.npy
├── phenotypes.hdf5
└── tasks
├── task0_LogisticTest
│ └── task_info.pkl
└── task1_LinearTest
└── task_info.pkl
This contains all the results and information needed to describe the experiment. The only missing thing for perfect reproducibility is a copy of the input files. Eventually, an opt-out feature will allow users to have automatic archiving of the input files.
Here is a description of all of the results files.
configuration.yamlThe configuration file that was used to generate these results.experiment_info.pklA Python pickle file contining experiment meta-data. See the following example for details{'build': 'GRCh37', 'configuration': 'sample_experiment/experiment.yaml', 'engine_url': 'sqlite:///ADCY9_forward/forward_database.db', 'name': 'ADCY9_forward', 'outcomes': [u'Infarctus', u'Valve', u'Angine', u'Diabete', u'BMI', u'BPSystolic', u'BPDiastolic', u'PC1', u'PC2', u'PC3', u'GenderFemale', u'Age'], 'phen_correlation': 'My_Experiment/phen_correlation_matrix.npy', 'phenotype_correlation_for_exclusion': 0.8, 'start_time': datetime.datetime(2015, 10, 4, 16, 15, 51, 680559), 'walltime': datetime.timedelta(0, 51, 395957)}
forward_database.dbA sqlite3 database containing all the results. Internally, Forward uses SQL Alchemy to create the database, this means that it will be easy to support more robust RDBMS without lots of changes to the codebase. A sample database will have the following tables:continuous_variablesresultsdiscrete_variablesvariableslinreg_resultsvariantsrelated_phenotypes_exclusions
See the documentation of
forward.experiment.ExperimentResultfor a full description of the schema for the results table.phen_correlation_matrix.npyA numpy binary file containing a correlation matrix for the outcomes. This is used to compute the exclusions based on related outcome correlation.phenotypes.hdf5A HDF5 binary file containing all the data from the phenotype database. This is used by the report to create graphics on the fly, before and after transformations.tasksThis is a subdirectory containing task metadata in the Pickle format.
This is the “low-level” alternative for browsing results from Forward
experiments. Alternatively, if you dislike the web-based report but still want
easy access to experiment results, you can use the
forward.backend.Backend class directly from your own Python script.